by James R. Edgar
Abstract
What is the current definition of an exosome?
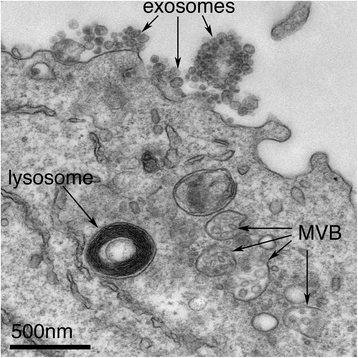
Exosomes correspond to intraluminal vesicles of multivesicular bodies. A transmission electron micrograph of an Epstein–Barr virus-transformed B cell displaying newly expelled exosomes at the plasma membrane. Multivesicular bodies (MVB) can be seen which can deliver content to lysosomes for degradation or can fuse with the cell surface to release intraluminal vesicles as exosomes, indicated by the arrows at the top of the picture
There are other types of microvesicle, including apoptotic bodies and ectosomes, which are derived from cells undergoing apoptosis and plasma membrane shedding, respectively. Although apoptotic bodies, ectosomes and exosomes are all roughly the same size (typically 40–100 nm) and all also contain ‘gulps’ of cytosol, they are different species of vesicles and understanding differences between them is of paramount importance but has too often been overlooked.
How were exosomes first recognized as distinct entities?
Even then, however, these extracellular vesicles were largely ignored, forgotten or, again, dismissed as a means of cellular waste disposal. It is only in the past decade that interest in exosomes has exploded, with a nearly tenfold increase in publications in as many years (115 in 2006, 1010 in 2015).
Why this explosion of interest?
Yet despite 20 years of research, the very basics of exosome biology are in their infancy and we know little of the part they play in normal cellular physiology.
So do we know how they are generated?
ILVs (and thus exosomes) can be generated at the endosomal limiting membrane by at least two mechanisms, one of which depends on the ESCRT machinery (ESCRT stands for endosomal sorting complexes required for transport) whereas the other is ESCRT-independent (Fig. 2).
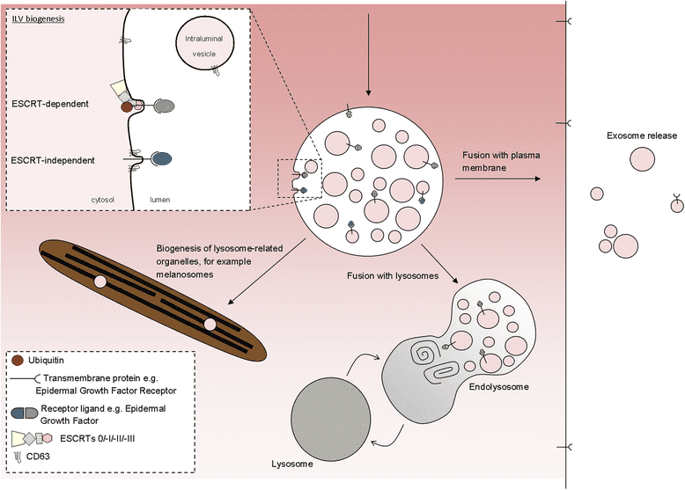
ILVs are generated by invagination of the endosomal membrane and have three possible fates. Inset: intraluminal vesicles (ILV) are formed by invagination of the endosomal membrane by either ESCRT-dependent or ESCRT-independent mechanisms. Matured endosomes accumulate ILVs within their lumen and have three distinct fates. They may deliver content that contributes to the biogenesis of specialized lysosome-related organelles (for example, melanosomes, Weibel-Palade bodies, azurophilic granules), they may fuse with lysosomes or they may fuse with the plasma membrane where released ILVs are now termed ‘exosomes’
But ILVs are still able to form in the absence of ESCRTs [8], so other means of generating ILVs must exist, although the mechanisms for their generation are less clear. Generation of these ESCRT-independent ILVs requires the tetraspanin CD63—a protein abundant on ILVs but with unclear function [9]—and may be facilitated by cone-shaped bending properties of lipids such as ceramide [10].
If not all ILVs become exosomes, what determines the fate of an ILV?
But what regulates the balance between exosome release and alternative fates of ILVs remains engimatic.
What about differences between cells: do all cells release exosomes?
Some cells—for example, the B cells, dendritic cells and mast cells of the immune system—appear to release exosomes constitutively; in fact, most of the data we have on exosomes comes from immune cells. As well as releasing exosomes constitutively, these cells may also be stimulated to secrete exosomes by cellular interactions. For example, murine dendritic cells, which are specialized to activate T lymphocytes, secrete higher levels of exosomes upon interaction with antigen-specific CD4+ T lymphocytes [13]. In fact, lymphocyte interactions generally can be accompanied by exosome release; human T cells (including primary T cells from blood, T cell clones and Jurkat cell lines) release exosomes upon activation of their antigen receptors [14] and B cells release more exosomes upon engagement with antigen-specific CD4+ T cells [15].
Other cell types can be pushed to secrete exosomes by means of calcium ionophores or other stimuli[16, 17], but the extent of physiological exosome secretion in non-immune cells is largely unknown.
What happens when exosomes reach an acceptor cell?
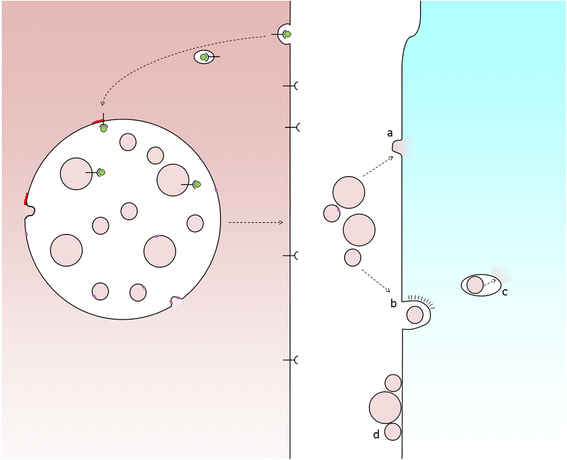
Exosome uptake by recipient cells. Fusion of MVBs with the cell surface releases ILVs as exosomes. In order for exosomes to elicit a response from recipient cells they might either fuse with plasma membrane (a) or be taken up whole via endocytosis (b), following which the exosome must be delivered to the cytosol, for example, via a back-fusion event (c). Alternatively, exosomes may attach to the surface of recipient cells to elicit a signalling response (d)
For intercellular transmission, various mechanisms of phagocytosis and endocytosis of extracellular vesicles have been described and which mechanism operates may depend upon vesicle size, which may in turn depend upon the cargo carried by the vesicle. In order for material to be released to an acceptor cell, exosomes must fuse with the host cell and this takes place via either direct fusion with the plasma membrane or a ‘back-fusion’ step from within a host endocytic organelle after the exosome has been engulfed. The process of back-fusion is not entirely clear, although it appears to require the unconventional lipid LBPA and protein Alix [19] (and is exploited by anthrax toxin lethal factor to escape from endosomes to the cytosol [20]).
Whether exosomes fuse with target cells or act via interactions with cell-surface proteins, or both, is another fundamental cell biology question that will need to be addressed if we are to understand the functions of exosomes.
So what are the consequences of all this information transfer? What biological functions have been established for exosomes?
Or, as with follicular dendritic cells, exosome-associated MHC II can be found on the surface of cell types that neither express MHC II nor secrete exosomes, indicating that exosomes are delivered from one cell type to another [18].
How exactly would exosomes from one cell influence the expression and activity of proteins in an acceptor cell?
So exosomes can also contribute to disease?
Of the neurodegenerative-associated proteins, only some are integral membrane proteins, that is, proteins inserted into lipid bilayers, rather than cytosolic. Sorting of proteins into ILVs (and thus exosomes) is easier to envisage for membrane proteins, where tags such as ubiquitin regulate where they end up. So far, the presence of both Aß [29] and PrPc [26] has in fact been shown in ILVs, though this has not been demonstrated for other membrane proteins, such as alpha-synuclein and tau.
The mechanism whereby cytosolic proteins may be sorted to ILVs/exosomes, however, is not clear. In order for cytosolic proteins to become concentrated in ILVs, they would require positive incorporation and sorting, possibly by membrane-associated components on endosomes. All we can say is that there is evidence that this does in fact happen; cytosolic factors such as miRNAs are enriched in exosomes relative to cytosol, indicating that sorting must occur whereby certain miRNAs are concentrated and others are not [30].
The means by which disease-associated factors spread between cells remains poorly understood and exosomes would provide a means for such transmission. The presence of exosomal proteins, such as Alix, in association with Alzheimer’s senile plaques strengthens the circumstantial case for exosomes as a mediator in such spread. The hope is that having a means to regulate exosome release and spread may be useful in combatting some of these diseases but much more basic biology needs to be established before then.
Now I’m confused—what determines what exosomes contain?
The mechanisms that concentrate cytosolic factors are currently unknown. Although it seems clear that miRNAs, for example, are enriched relative to the amount in their parent cells, and are not randomly incorporated into exosomes, it is not clear how some are enriched more than others. There are currently a few hypotheses for miRNA sorting, including sorting via sumoylated heterogeneous nuclear ribonucleoproteins [31] or by a miRNA-induced silencing complex (miRISC) [32].
Because of the difficulties in separating exosomes from other extracellular vesicles, it is likely that some cargos reported to be enriched in ‘exosomes’ may in fact be contained in contaminant vesicles that are not exosomes. While many researchers are very stringent about applying the labels ‘exosomes’ and ‘extracellular vesicles’ correctly, others unfortunately are not. In addition, as I have said before, cytosolic proteins are likely to be found in exosome preparations because the exosome lumen is made of cytosol.
So how exactly can you be sure that a given extracellular vesicle is an exosome and not something else?
One problem is that ILVs, and thus exosomes, represent an intermediate compartment of an intermediate. MVBs are not static organelles but rather undergo continuous maturation, in the course of which they gain and lose proteins. There will never be an exclusive marker for exosomes because any cargo on the ILV/exosome membrane must first be on the limiting membrane of the endosome and anything found inside must first come from the cytosol. A cargo may be concentrated on ILVs/exosomes but it will also be elsewhere. CD63 could be thought of as a pseudo-marker for exosomes. ILVs and exosomes are enriched in several such tetraspanins and my colleagues and I have show that CD63 is required for ESCRT-independent ILV formation [9]. Alix also appears to be concentrated in ILVs/exosomes [33], as does Tsg101, a component of ESCRT-I, which has been used as a marker of exosomes in numerous studies [33,34], although the presence of Tsg101 in ILVs or exosomes does not fit with conventional models of ILV formation. Although Tsg101 is involved in ESCRT-dependent ILV formation, as mentioned earlier, it, along with other ESCRT components, should disassociate from the endosomal membrane prior to an ILV pinching off the endosomal membrane to allow it to participate in further events [35]. Exactly when ESCRT-I components ‘fall off’ the membrane is unknown but it is conventionally thought to be prior to ILV formation, so Tsg101 should remain cytosolic and available for subsequent rounds of ILV formation. It is possible that some Tsg101 may be ‘swallowed’ into the forming ILV lumen, but levels should be negligible.
So are you saying there is no reliable marker for endosomes?
If they are so hard to characterize reliably, how are exosomes isolated and studied?
What would you say are the most important issues in exosome research?
References
-
Harding C, Heuser J, Stahl P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J Cell Biol. 1983;97(2):329–39.
-
Pan BT, Teng K, Wu C, Adam M, Johnstone RM. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J Cell Biol. 1985;101(3):942–8.
-
Johnstone RM, Adam M, Hammond JR, Orr L, Turbide C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J Biol Chem. 1987;262(19):9412–20.
-
Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, et al. B lymphocytes secrete antigen-presenting vesicles. J Exp Med. 1996;183(3):1161–72.
-
Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza D, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med. 1998;4(5):594–600.
-
Kim MS, Haney MJ, Zhao Y, Mahajan V, Deygen I, Klyachko NL, et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomedicine. 2015;12(3):655–64.
-
McDonald B, Martin-Serrano J. No strings attached: the ESCRT machinery in viral budding and cytokinesis. J Cell Sci. 2009;122(Pt 13):2167–77.
-
Stuffers S, Sem Wegner C, Stenmark H, Brech A. Multivesicular endosome biogenesis in the absence of ESCRTs. Traffic. 2009;10(7):925–37.
-
Edgar JR, Eden ER, Futter CE. Hrs- and CD63-dependent competing mechanisms make different sized endosomal intraluminal vesicles. Traffic. 2014;15(2):197–211.
-
Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008;319(5867):1244–7.
-
White IJ, Bailey LM, Aghakhani MR, Moss SE, Futter CE. EGF stimulates annexin 1-dependent inward vesiculation in a multivesicular endosome subpopulation. EMBO J. 2006;25(1):1–12.
-
Mobius W, Ohno-Iwashita Y, van Donselaar EG, Oorschot VM, Shimada Y, Fujimoto T, et al. Immunoelectron microscopic localization of cholesterol using biotinylated and non-cytolytic perfringolysin O. J Histochem Cytochem. 2002;50(1):43–55.
-
Buschow SI, Nolte-’t Hoen EN, van Niel G, Pols MS, ten Broeke T, Lauwen M, et al. MHC II in dendritic cells is targeted to lysosomes or T cell-induced exosomes via distinct multivesicular body pathways. Traffic. 2009;10(10):1528–42.
-
Blanchard N, Lankar D, Faure F, Regnault A, Dumont C, Raposo G, et al. TCR activation of human T cells induces the production of exosomes bearing the TCR/CD3/zeta complex. J Immunol. 2002;168(7):3235–41.
-
Muntasell A, Berger AC, Roche PA. T cell-induced secretion of MHC class II-peptide complexes on B cell exosomes. EMBO J. 2007;26(19):4263–72.
-
Savina A, Furlan M, Vidal M, Colombo MI. Exosome release is regulated by a calcium-dependent mechanism in K562 cells. J Biol Chem. 2003;278(22):20083–90.
-
Guo BB, Bellingham SA, Hill AF. Stimulating the release of exosomes increases the intercellular transfer of prions. J Biol Chem. 2016;291(10):5128–37.
-
Denzer K, van Eijk M, Kleijmeer MJ, Jakobson E, de Groot C, Geuze HJ. Follicular dendritic cells carry MHC class II-expressing microvesicles at their surface. J Immunol. 2000;165(3):1259–65.
-
Bissig C, Gruenberg J. ALIX and the multivesicular endosome: ALIX in Wonderland. Trends Cell Biol. 2014;24(1):19–25.
-
Abrami L, Lindsay M, Parton RG, Leppla SH, van der Goot FG. Membrane insertion of anthrax protective antigen and cytoplasmic delivery of lethal factor occur at different stages of the endocytic pathway. J Cell Biol. 2004;166(5):645–51.
-
Lo Cicero A, Delevoye C, Gilles-Marsens F, Loew D, Dingli F, Guere C, et al. Exosomes released by keratinocytes modulate melanocyte pigmentation. Nat Commun. 2015;6:7506.
-
Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–9.
-
Melo SA, Sugimoto H, O’Connell JT, Kato N, Villanueva A, Vidal A, et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell. 2014;26(5):707–21.
-
Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, et al. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A. 2006;103(30):11172–7.
-
Saman S, Kim W, Raya M, Visnick Y, Miro S, Saman S, et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem. 2012;287(6):3842–9.
-
Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, et al. Cells release prions in association with exosomes. Proc Natl Acad Sci U S A. 2004;101(26):9683–8.
-
Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, et al. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30(20):6838–51.
-
Gomes C, Keller S, Altevogt P, Costa J. Evidence for secretion of Cu, Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neurosci Lett. 2007;428(1):43–6.
-
Guduric-Fuchs J, O’Connor A, Camp B, O’Neill CL, Medina RJ, Simpson DA. Selective extracellular vesicle-mediated export of an overlapping set of microRNAs from multiple cell types. BMC Genomics. 2012;13:357.
-
Edgar JR, Willen K, Gouras GK, Futter CE. ESCRTs regulate amyloid precursor protein sorting in multivesicular bodies and intracellular amyloid-beta accumulation. J Cell Sci. 2015;128(14):2520–8.
-
Villarroya-Beltri C, Gutierrez-Vazquez C, Sanchez-Cabo F, Perez-Hernandez D, Vazquez J, Martin-Cofreces N, et al. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun. 2013;4:2980.
-
Fabian MR, Sonenberg N. The mechanics of miRNA-mediated gene silencing: a look under the hood of miRISC. Nat Struct Mol Biol. 2012;19(6):586–93.
-
Bobrie A, Colombo M, Krumeich S, Raposo G, Thery C. Diverse subpopulations of vesicles secreted by different intracellular mechanisms are present in exosome preparations obtained by differential ultracentrifugation. J Extracell Vesicles. 2012;1:18397.
-
Thery C, Boussac M, Veron P, Ricciardi-Castagnoli P, Raposo G, Garin J, et al. Proteomic analysis of dendritic cell-derived exosomes: a secreted subcellular compartment distinct from apoptotic vesicles. J Immunol. 2001;166(12):7309–18.
-
Hurley JH. The ESCRT complexes. Crit Rev Biochem Mol Biol. 2010;45(6):463–87.
-
ExoCarta. http://www.exocarta.org.
-
Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protocols Cell Biol. 2006;Chapter 3:Unit 3 22.
-
Lotvall J, Hill AF, Hochberg F, Buzas EI, Di Vizio D, Gardiner C, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles. 2014;3:26913.
Acknowledgements
Competing interests
The author declares that he has no competing interests.






